| Experiments with Serial NetEvolve | |
|
Patricia
Buendia and Giri Narasimhan
|
|
Supplemental website for manuscript titled "Serial NetEvolve: A flexible utility for generating serially-sampled sequences along a tree or recombinant network" |
|
| The unique features of Serial NetEvolve allowed us to perform with relative ease a comparison study of tree topologies output by several methods on synthetically generated data sets. Here we briefly present the results of two comparison studies. The first study compares topologies and ancestor/descendant relationships inferred by six methods under the two settings of the molecular clock. The second study compares four methods under different sampling strategies. | |
Clock Study |
|
| Our
comparison study consisted in evaluating five methods for their accuracy
in inferring the true topology from simulated data generated by Serial
NetEvolve using the Symmetric
Difference Score of Robinson and Foulds (Robinson and Foulds 1981).
We also evaluated the methods under the A-D branch length score: a
measure based
on the percentage of correctly inferred ancestor-descendant
relationships (Buendia et al.
2006). For
a given descendant taxon, the closest ancestor is defined as the closest
sequence (i.e., with minimum branch length distance) sampled at some
previous sampling time. In particular, we investigated how the molecular clock
hypothesis affected their relative performance, as two of the algorithms
that accept serially-sampled data as input, TipDate (Rambaut
2000)
and sUPGMA (Drummond and Rodrigo 2000),
assume a molecular clock. TipDate does not infer a tree, but requires a
known topology as part of its input and it re-computes the branch
lengths to fit the molecular clock. For the input tree topology to
TipDate, we used the topology estimated by the fastDNAml method. Two
other methods for serially sampled data were included in the study,
MinPD (Buendia and Narasimhan
2004) and
SeqLink (Ren
et al. 2003). Two standard phylogenetic programs were also included:
fastDNAml (Olsen et al. 1994)and DNAPARS (Felsenstein 2004).
1000 replicates were generated under the following settings: Sequence length of 1000, no internal nodes sampling, recombination rate of zero, exponential rate of 0.0001, sample size per time point = 8, sampling times = 6, mutation rate = 10-5, population size = 106, model = HKY with rate heterogeneity, Ts/Tv = 4, and alpha parameter = 0.5. |
|
 |
|
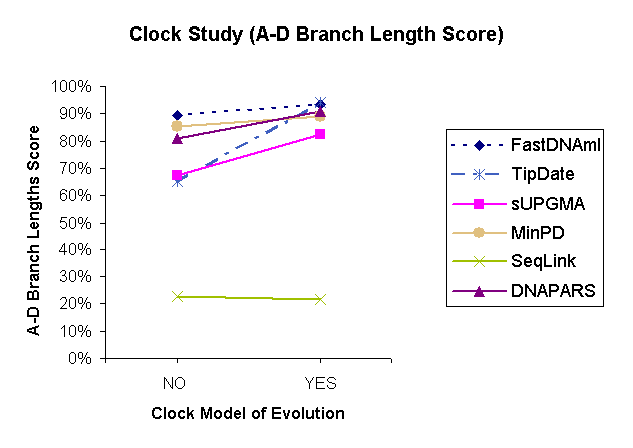
| Figure 1: Results of a comparison study of 6 methods under the Ancestor-Descendant Score based on the variable parameter "Molecular clock." | |
 |
|
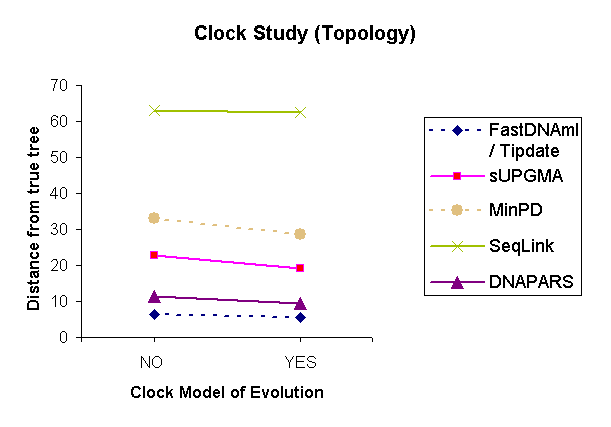
| Figure 2: Results of a topology comparison study of 6 methods based on the variable parameter "Molecular clock." | |
Sampling Strategies Study |
|
| In this study the
standard phylogenetic methods Neighbor Joining and UPGMA were compared
with two other methods designed to analyze serially-sampled data, sUPGMA
(Drummond and Rodrigo 2000) and MinPD (Buendia and Narasimhan 2004). The
Serial Coalescent Simulator as described in (Drummond and Rodrigo 2000)
was used to test the performance of sUPGMA and UPGMA with serial samples
evolved under variable inter-sample divergences. In the Serial
NetEvolve study the symmetrical interval distance between sampling
points was used instead of the inter-sample divergence, as it is not a
parameter of Serial NetEvolve. We present results for 1000
replicates with samples of size 20, 40, and 80 and a range of interval
distances between 100 and 2600. The graphs show the performance of the 4
programs using different interval distances evaluated by the topological
distance measure (Robinson and Foulds 1981).
Non-default settings were: The JC Model of evolution, no site-specific rate heterogeneity, mutation rate of 5x10-6, sequence length of 1000, no internal nodes sampling, the clock model, and a recombination rate of zero. Coalescent Trees were generated under different sampling strategies and with 4 sampling times and variable samples sizes of 5, 10 and 20 per sampling time. |
|
 |
|
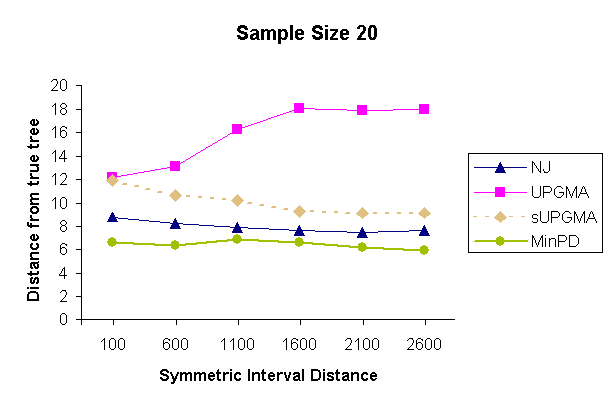
| Figure 3: Results of a topology comparison study of 4 methods with a sampling strategy of 5 samples per sampling time and 4 sampling times. | |
 |
|
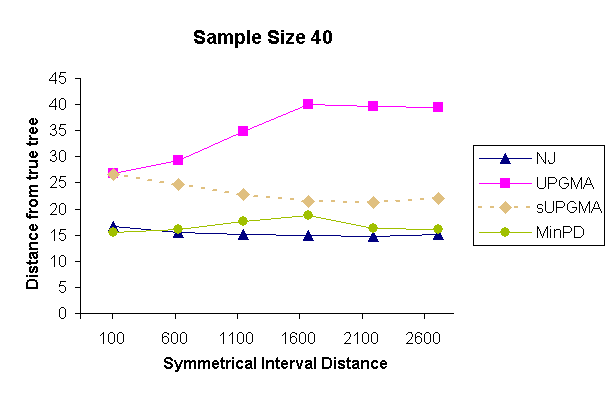
| Figure 4: Results of a topology comparison study of 4 methods with a sampling strategy of 10 samples per sampling time and 4 sampling times. | |
 |
|
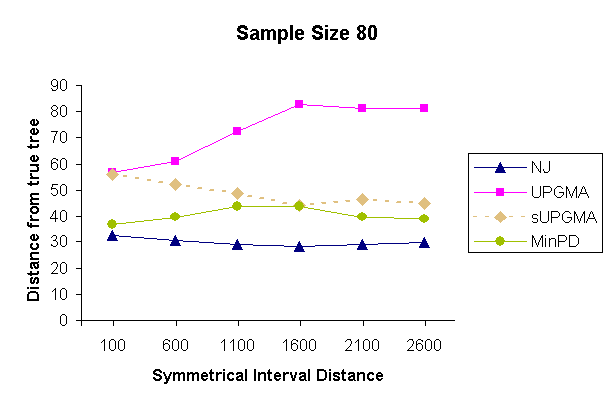
| Figure 5: Results of a topology comparison study of 4 methods with a sampling strategy of 20 samples per sampling time and 4 sampling times. | |
References |
|
| Buendia, P. and G.
Narasimhan. (2005). MinPD: Distance-based Phylogenetic Analysis and
Recombination Detection of Serially-Sampled HIV Quasispecies. Proc. IEEE
Comput. Sys. Bioinform. Conf., Stanford, CA
Buendia, P., Collins, T., and Narasimhan, G. (2006). Reconstructing Ancestor-Descendant Lineages from Serially-Sampled Data: A Comparison Study. International Conference on Computational Science (IWBRA06), Reading, UK. 807-814. Drummond, A. and A. G. Rodrigo. (2000). Reconstructing genealogies of serial samples under the assumption of a molecular clock using serial-sample UPGMA (sUPGMA). Mol. Biol. Evol. 17:1807-1815. Felsenstein, J. 2004. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genetics, University of Washington, Seattle Olsen, G. J., et al. 1994. fastDNAml: A tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput. Appl. Biosci. 10:41-48. Rambaut, A. 2000. Estimating the rate of molecular evolution: Incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 16:395-399. Ren, F., et al. 2003. Longitudinal phylogenetic tree of within-host viral evolution from non-contemporaneous samples: a distance-based sequential-linking method. Gene 317(1-2):89-95. Robinson, D. F. and L. R. Foulds. 1981. Comparison of phylogenetic trees. Mathematical Biosciences 53:131-147. |
|